2024-05-20
作者:刘东鑫,黄飞,李亚茹,毛凌峰,贺文从,吴斯豪,夏辉,何萍,郑慧文,周杨,赵冰,欧喜超,宋媛媛,宋泽萱,梅蠡,刘丽,张国良,魏强,赵雁林
研究背景:
中国的结核病负担位居全球第三,然而在全国范围内,影响结核病和MDR/RR-TB传播和出现的因素仍然研究不足。全基因组测序(WGS)显著提高了分离株之间遗传相关性的确定和耐药性的识别。本研究系统地收集和测序了来自全国31个省份70个县的5052个MTB分离株,以及相关流行病学信息。旨在利用这一具有全国代表性的数据集,评估中国基因组聚集性结核病的传播情况,并确定相关的保护/风险因素。同时,我们评估了不同簇中耐药突变的分布,并结合治疗史,量化了由传播引起的耐药性结核病的程度。基于本研究,我们希望制定有效的干预措施,以阻断结核病的传播并促进结核病的控制,主要目的是预防和减少MDR/RR结核病的出现。
研究方法:
本研究以全国70个耐药监测点的结核病人群数据为基础,收集了2013年全国范围内5052例患者的流行病学数据和临床菌株全基因组数据。基于全基因组测序数据预测了菌株对利福平和异烟肼的敏感性,并以12个SNPs阈值鉴定成簇;将簇内具有相同耐药突变的病例以及所有初治耐药病例定义为传播性耐药。确定了我国结核菌及耐药结核菌成簇情况,并使用单变量和多变量逻辑回归模型计算与基因组成簇相关的风险因素。
研究结果
我们鉴定了452个基因组簇,大小从2个到14个菌株不等,涉及5052个菌株中的1160个,在全国范围内的聚集比例为23.0%。387个簇(85.6%)中病例中菌株均来自同一县。65个跨县集群由2至7个隔离株组成,地理分布从2至3个县不等(图1)。采用有序logistic回归分析表明成簇风险因素包括年龄偏小(P-trend=0.01)、家庭规模偏大(调整后OR= 1.11 (95% CI, 1.07-1.15), P<0.001)、新发结核病例(aOR=1.48 (95% CI, 1.22-1.79), P<0.001)、痰涂片阳性(aOR=1.52 (95% CI, 1.26-1.83), P<0.001)、MDR/RR (aOR=1.59 (95% CI, 1.23-2.05), P<0.001)、高等教育(OR=0.57 (95% CI, 0.37-0.88),P=0.011)和职业为非体力工人(aOR=0.72 (95% CI, 0.54-0.95)、P=0.022)为保护因素。我们还发现429株MDR/RR菌株,其中121株与28株利福平敏感菌株聚在50个基因簇中(图2)。基于基因簇和治疗史我们确定至少61.4%(251/409)的MDR/RR结核患者可能是由最近在整个中国传播的MDR/RR菌株引起的(图3)。

图1我国结核分枝杆菌成簇情况
注:12株snp株的遗传距离定义为集群,仅集群菌株被保留。每个圆代表一个集群。圆圈的大小表示簇中应变的数量。每个圆圈的颜色代表菌株的监测地点。从同一组内同一县分离的菌株呈白色,但不代表所有白色菌株均从同一位点分离的
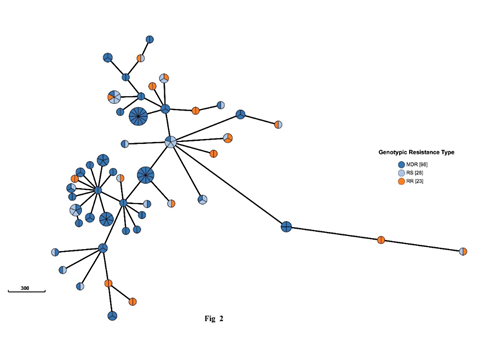
图2多药耐药或利福平耐药菌株与利福平敏感菌株的基因簇
注:98株耐多药(MDR)株、23株利福平耐药(RR)株和28株利福平敏感(RS)株聚集在50个WGS-簇中。
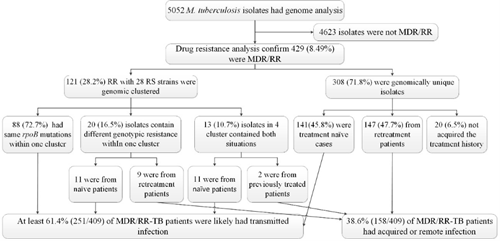
图3根据治疗史和基因组分析对MDR/RR-TB来源的分类
研究结论及意义
我们采用联合基因组流行病学方法评估了中国结核病和MDR/RR-TB在人群水平上的聚类特征,并分析了与近期传播相关的危险因素。此外,我们证明耐药菌株的传播在整个中国的MDR/RR-TB负担中发挥着重要作用。本研究的实施为制定遏制中国结核病和耐药结核病流行措施的制定提供了理论基础,结果表明当前我国迫切需要早期诊断结核病、快速诊断耐药以及对高危人群和耐药患者进行严格隔离治疗等干预措施。此外,以当前基因组数据库为基础,通过研究人员不断提交增加数据库可以监测我国不同区域间测结核病传播情况。
本研究成果发表在Emerg Microbes Infect. 2024 Apr 30:2348505. doi:10.1080/22221751.2024.2348505.